VALIDATION AND QUALIFICATION OF WATER PURIFICATION, STORAGE, AND DISTRIBUTION SYSTEMS: Establishing the dependability of pharmaceutical water purification, storage, and distribution systems requires an appropriate period of monitoring and observation. Ordinarily, few problems are encountered in maintaining the chemical purity of Purified Water and Water for Injection Nevertheless, the advent of using conductivity and TOC to define chemical purity has allowed the user to more quantitatively assess the water’s chemical purity and its variability as a function of routine pretreatment system maintenance and regeneration. Even the presence of such unit operations as heat exchangers and use point hoses can compromise the chemical quality of water within and delivered from an otherwise well-controlled water system. Therefore, an assessment of the consistency of the water’s chemical purity over time must be part of the validation program. However, even with the most well controlled chemical quality, it is often more difficult to consistently meet established microbiological quality criteria owing to phenomena occurring during and after chemical purification. A typical program involves intensive daily sampling and testing of major process points for at least one month after operational criteria have been established for each unit operation, point of use, and sampling point.
VALIDATION AND QUALIFICATION OF WATER PURIFICATION, STORAGE, AND DISTRIBUTION SYSTEMS
An overlooked aspect of water system validation is the delivery of the water to its actual location of use. If this transfer process from the distribution system outlets to the water use locations (usually with hoses) is defined as outside the water system, then this transfer process still needs to be validated to not adversely affect the quality of the water to the extent it becomes unfit for use. Because routine microbial monitoring is performed for the same transfer process and components (e.g., hoses and heat exchangers) as that of routine water use (see Sampling Considerations), there is some logic to include this water transfer process within the distribution system validation.
Validation is the process whereby substantiation to a high level of assurance that a specific process will consistently produce a product conforming to an established set of quality attributes is acquired and documented. Prior to and during the very early stages of validation, the critical process parameters and their operating ranges are established. A validation program qualifies and documents the design, installation, operation, and performance of equipment. It begins when the system is defined and moves through several stages: installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ). A graphical representation of a typical water system validation life cycle is shown in Figure.
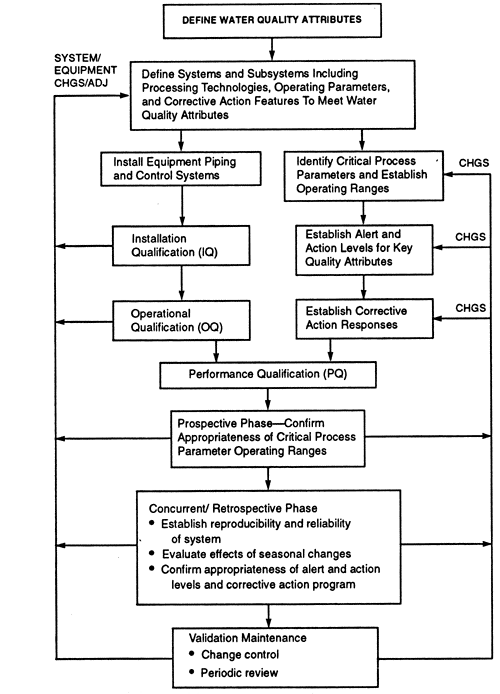
Pharmaceutical Water system validation life cycle.
A validation plan for a water system typically includes the following steps: (1) establishing standards for quality attributes of the finished water and the source water; (2) defining suitable unit operations and their operating parameters for achieving the desired finished water quality attributes from the available source water; (3) selecting piping, equipment, controls, and monitoring technologies; (4) developing an IQ stage consisting of instrument calibrations, inspections to verify that the drawings accurately depict the final configuration of the water system and, where necessary, special tests to verify that the installation meets the design requirements; (5) developing an OQ stage consisting of tests and inspections to verify that the equipment, system alerts, and controls are operating reliably and that appropriate alert and action levels are established (This phase of qualification may overlap with aspects of the next step.); and (6) developing a prospective PQ stage to confirm the appropriateness of critical process parameter operating ranges (During this phase of validation, alert and action levels for key quality attributes and operating parameters are verified.); (7) assuring the adequacy of ongoing control procedures, e.g., sanitization frequency; (8) supplementing a validation maintenance program (also called continuous validation life cycle) that includes a mechanism to control changes to the water system and establishes and carries out scheduled preventive maintenance including recalibration of instruments (In addition, validation maintenance includes a monitoring program for critical process parameters and a corrective action program.); (9) instituting a schedule for periodic review of the system performance and requalification, and (10) completing protocols and documenting Steps 1 through 9.
PURIFIED WATER AND WATER FOR INJECTION SYSTEMS
The design, installation, and operation of systems to produce Purified Water and Water for Injection include similar components, control techniques, and procedures. The quality attributes of both waters differ only in the presence of a bacterial endotoxin requirement for Water for Injection and in their methods of preparation, at least at the last stage of preparation. The similarities in the quality attributes provide considerable common ground in the design of water systems to meet either requirement. The critical difference is the degree of control of the system and the final purification steps needed to ensure bacterial and bacterial endotoxin removal.
Production of pharmaceutical water
Production of pharmaceutical water employs sequential unit operations (processing steps) that address specific water quality attributes and protect the operation of subsequent treatment steps. A typical evaluation process to select an appropriate water quality for a particular pharmaceutical purpose is shown in the decision tree in Figure 2. This diagram may be used to assist in defining requirements for specific water uses and in the selection of unit operations. The final unit operation used to produce Water for Injection is limited to distillation or other processes equivalent or superior to distillation in the removal of chemical impurities as well as microorganisms and their components. Distillation has a long history of reliable performance and can be validated as a unit operation for the production of Water for Injection, but other technologies or combinations of technologies can be validated as being equivalently effective. Other technologies, such as ultrafiltration following other chemical purification process, may be suitable in the production of Water for Injection if they can be shown through validation to be as effective and reliable as distillation. The advent of new materials for older technologies, such as reverse osmosis and ultrafiltration, that allow intermittent or continuous operation at elevated, microbial temperatures, show promise for a valid use in producing Water for Injection.
[PPT PDF] Pharmaceutical Water System Design Operation And Validation Pdf PowerPoint Pharmaceutical Water System Design Operation And Validation Pdf PowerPoint
Validation plan Pharmaceutical Water System
The validation plan should be designed to establish the suitability of the system and to provide a thorough understanding of the purification mechanism, range of operating conditions, required pretreatment, and the most likely modes of failure. It is also necessary to demonstrate the effectiveness of the monitoring scheme and to establish the documentation and qualification requirements for the system’s validation maintenance. Trials conducted in a pilot installation can be valuable in defining the operating parameters and the expected water quality and in identifying failure modes. However, qualification of the specific unit operation can only be performed as part of the validation of the installed operational system. The selection of specific unit operations and design characteristics for a water system should take into account the quality of the feed water, the technology chosen for subsequent processing steps, the extent and complexity of the water distribution system, and the appropriate compendial requirements. For example, in the design of a system for Water for Injection, the final process (distillation or whatever other validated process is used according to the monograph) must have effective bacterial endotoxin reduction capability and must be validated.
INSTALLATION, MATERIALS OF CONSTRUCTION, AND COMPONENT SELECTION
Installation techniques are important because they can affect the mechanical, corrosive, and sanitary integrity of the system. Valve installation attitude should promote gravity drainage. Pipe supports should provide appropriate slopes for drainage and should be designed to support the piping adequately under worst-case thermal and flow conditions. The methods of connecting system components including units of operation, tanks, and distribution piping require careful attention to preclude potential problems. Stainless steel welds should provide reliable joints that are internally smooth and corrosion-free. Low-carbon stainless steel, compatible wire filler, where necessary, inert gas, automatic welding machines, and regular inspection and documentation help to ensure acceptable weld quality. Follow-up cleaning and passivation are important for removing contamination and corrosion products and to re-establish the passive corrosion resistant surface. Plastic materials can be fused (welded) in some cases and also require smooth, uniform internal surfaces. Adhesive glues and solvents should be avoided due to the potential for voids and extractables. Mechanical methods of joining, such as flange fittings, require care to avoid the creation of offsets, gaps, penetrations, and voids. Control measures include good alignment, properly sized gaskets, appropriate spacing, uniform sealing force, and the avoidance of threaded fittings.
Materials of construction should be selected to be compatible with control measures such as sanitizing, cleaning, and passivating. Temperature rating is a critical factor in choosing appropriate materials because surfaces may be required to handle elevated operating and sanitization temperatures. Should chemicals or additives be used to clean, control, or sanitize the system, materials resistant to these chemicals or additives must be utilized. Materials should be capable of handling turbulent flow and elevated velocities without wear of the corrosion-resistant film such as the passive chromium oxide surface of stainless steel. The finish on metallic materials such as stainless steel, whether it is a refined mill finish, polished to a specific grit, or an electropolished treatment, should complement system design and provide satisfactory corrosion and microbial activity resistance as well as chemical sanitizability. Auxiliary equipment and fittings that require seals, gaskets, diaphragms, filter media, and membranes should exclude materials that permit the possibility of extractables, shedding, and microbial activity. Insulating materials exposed to stainless steel surfaces should be free of chlorides to avoid the phenomenon of stress corrosion cracking that can lead to system contamination and the destruction of tanks and critical system components.
Specifications are important to ensure proper selection of materials and to serve as a reference for system qualification and maintenance. Information such as mill reports for stainless steel and reports of composition, ratings, and material handling capabilities for nonmetallic substances should be reviewed for suitability and retained for reference. Component (auxiliary equipment) selection should be made with assurance that it does not create a source of contamination intrusion. Heat exchangers should be constructed to prevent leakage of heat transfer medium to the pharmaceutical water and, for heat exchanger designs where prevention may fail, there should be a means to detect leakage. Pumps should be of sanitary design with seals that prevent contamination of the water. Valves should have smooth internal surfaces with the seat and closing device exposed to the flushing action of water, such as occurs in diaphragm valves. Valves with pocket areas or closing devices (e.g., ball, plug, gate, globe) that move into and out of the flow area should be avoided.
SANITIZATION – Pharmaceutical Water System
Microbial control in water systems is achieved primarily through sanitization practices. Systems can be sanitized using either thermal or chemical means. Thermal approaches to system sanitization include periodic or continuously circulating hot water and the use of steam. Temperatures of at least 80 are most commonly used for this purpose, but continuously recirculating water of at least 65 has also been used effectively in insulated stainless steel distribution systems when attention is paid to uniformity and distribution of such self-sanitizing temperatures. These techniques are limited to systems that are compatible with the higher temperatures needed to achieve sanitization. Although thermal methods control biofilm development by either continuously inhibiting their growth or, in intermittent applications, by killing the microorganisms within biofilms, they are not effective in removing established biofilms. Killed but intact biofilms can become a nutrient source for rapid biofilm regrowth after the sanitizing conditions are removed or halted. In such cases, a combination of routine thermal and periodic supplementation with chemical sanitization might be more effective. The more frequent the thermal sanitization, the more likely biofilm development and regrowth can be eliminated. Chemical methods, where compatible, can be used on a wider variety of construction materials. These methods typically employ oxidizing agents such as halogenated compounds, hydrogen peroxide, ozone, peracetic acid, or combinations thereof. Halogenated compounds are effective sanitizers but are difficult to flush from the system and may leave biofilms intact. Compounds such as hydrogen peroxide, ozone, and peracetic acid oxidize bacteria and biofilms by forming reactive peroxides and free radicals (notably hydroxyl radicals). The short half-life of ozone in particular, and its limitation on achievable concentrations require that it be added continuously during the sanitization process. Hydrogen peroxide and ozone rapidly degrade to water and oxygen; peracetic acid degrades to acetic acid in the presence of UV light. In fact, ozone’s ease of degradation to oxygen using 254-nm UV lights at use points allow it to be most effectively used on a continuous basis to provide continuously sanitizing conditions.
In-line UV light at a wavelength of 254 nm can also be used to continuously “sanitize” water circulating in the system, but these devices must be properly sized for the water flow. Such devices inactivate a high percentage (but not 100%) of microorganisms that flow through the device but cannot be used to directly control existing biofilm upstream or downstream of the device. However, when coupled with conventional thermal or chemical sanitization technologies or located immediately upstream of a microbially retentive filter, it is most effective and can prolong the interval between system sanitizations.
It is important to note that microorganisms in a well-developed biofilm can be extremely difficult to kill, even by aggressive oxidizing biocides. The less developed and therefore thinner the biofilm, the more effective the biocidal action. Therefore, optimal biocide control is achieved by frequent biocide use that does not allow significant biofilm development between treatments.
Sanitization steps require validation to demonstrate the capability of reducing and holding microbial contamination at acceptable levels. Validation of thermal methods should include a heat distribution study to demonstrate that sanitization temperatures are achieved throughout the system, including the body of use point valves. Validation of chemical methods require demonstrating adequate chemical concentrations throughout the system, exposure to all wetted surfaces, including the body of use point valves, and complete removal of the sanitant from the system at the completion of treatment. Methods validation for the detection and quantification of residues of the sanitant or its objectionable degradants is an essential part of the validation program. The frequency of sanitization should be supported by, if not triggered by, the results of system microbial monitoring. Conclusions derived from trend analysis of the microbiological data should be used as the alert mechanism for maintenance.The frequency of sanitization should be established in such a way that the system operates in a state of microbiological control and does not routinely exceed alert levels (see Alert and Action Levels and Specifications).
Pharmaceutical Water System OPERATION, MAINTENANCE, AND CONTROL
A preventive maintenance program should be established to ensure that the water system remains in a state of control. The program should include (1) procedures for operating the system, (2) monitoring programs for critical quality attributes and operating conditions including calibration of critical instruments, (3) schedule for periodic sanitization, (4) preventive maintenance of components, and (5) control of changes to the mechanical system and to operating conditions.
Operating Procedures—
Procedures for operating the water system and performing routine maintenance and corrective action should be written, and they should also define the point when action is required. The procedures should be well documented, detail the function of each job, assign who is responsible for performing the work, and describe how the job is to be conducted. The effectiveness of these procedures should be assessed during water system validation.
Monitoring Program—
Critical quality attributes and operating parameters should be documented and monitored. The program may include a combination of in-line sensors or automated instruments (e.g., for TOC, conductivity, hardness, and chlorine), automated or manual documentation of operational parameters (such as flow rates or pressure drop across a carbon bed, filter, or RO unit), and laboratory tests (e.g., total microbial counts). The frequency of sampling, the requirement for evaluating test results, and the necessity for initiating corrective action should be included.
Sanitization—
Depending on system design and the selected units of operation, routine periodic sanitization may be necessary to maintain the system in a state of microbial control. Technologies for sanitization are described above.
Preventive Maintenance—
A preventive maintenance program should be in effect. The program should establish what preventive maintenance is to be performed, the frequency of maintenance work, and how the work should be documented.
Change Control—
The mechanical configuration and operating conditions must be controlled. Proposed changes should be evaluated for their impact on the whole system. The need to requalify the system after changes are made should be determined. Following a decision to modify a water system, the affected drawings, manuals, and procedures should be revised.
Auxiliary Information— Staff Liaison : Gary E. Ritchie, M.Sc., Scientific Fellow
Expert Committee : (PW05) Pharmaceutical Waters 05
USP29–NF24 Page 3056
Pharmacopeial Forum : Volume No. 30(5) Page 1744
Phone Number : 1-301-816-8353
Pharmaceutical Water System Ppt
Pharmaceutical Water Systems
Purified Water Specification As Per Usp
Pharmaceutical Water System Design Operation And Validation Pdf
pharmaceutical water system design operation and validation
pharmaceutical water system ppt – What is Pharmaceutical water
purified water & Water for Injection SOP as per usp
Pharmaceutical Water Systems: Storage & Distribution Systems
pharmaceutical water system : Inspection of Pharmaceutical water systems
pharmaceutical water Production : Water purification systems
Pharmaceutical Water Systems: Types: Water quality specifications
Pharmaceutical Water System: principles for pharmaceutical water systems