Effective process validation contributes significantly to assuring drug quality. The basic principle of quality assurance is that a drug should be produced that is fit for its intended use.
This principle incorporates the understanding that the following conditions exist:
• Quality, safety, and efficacy are designed or built into the product.
• Quality cannot be adequately assured merely by in-process and finished-product inspection or testing.
As we have discussed effective process validation contributes significantly to assuring drug quality. The basic principle of quality assurance is that a drug should be produced that is fit for its intended use. Pharmaceutical Process Validation Protocol & Report Format Example PPT PDF is given here for autoclave and sterilization. First let us know what is Pharmaceutical Process Validation. Validation refers to establishing documented evidence that a process or system, when operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its pre-determined specifications and quality attributes. It is mandatory to have a system stock list put in place, the appropriate SOPs in place, and additionally to check the critical techniques and their documentation. Having a powerful efficient Computer System Validation System put in place will help ensure the stability of the electronic documents, allocate resources better and subsequently can yield long run cost discounts to the company.
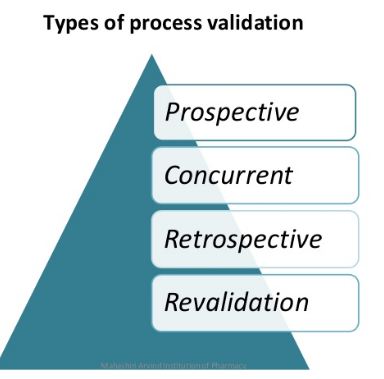
Approach to Process Validation:
For purposes of this guidance, process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific
evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the product and process. This
guidance describes process validation activities in three stages.
• Stage 1 – Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities.
• Stage 2 – Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
• Stage 3 – Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control.
Validation Protocol:
A written plan stating how validation will be conducted, including test parameters, product characteristics, production and packaging equipment, and decision points on what constitutes acceptable test results. This document should give details of critical steps of the manufacturing process that should be measured, the allowable range of variability and the manner in which the system will be tested.
The validation protocol provides a synopsis of what is hoped to be accomplished. The protocol should list the selected process and control parameters, state the number of batches to be included in the study, and specify how the data, once assembled, will be treated for relevance. The date of approval by the validation team should also be noted.
In the case where a protocol is altered or modified after its approval, appropriate reasoning for such a change must be documented.
The validation protocol should be numbered, signed and dated, and should contain as a minimum the following information:
1. Title
2. Objective & Scope
3. Responsibility
4. Protocol Approval
5. Validation Team
6. Product Composition
7. Process Flow Chart
8. Manufacturing Process
9. Review of Equipments / Utilities
10.Review of Raw Materials and Packing Materials
11. Review of Analytical and Batch Manufacturing Records
12. Review of Batch Quantities for Validation (Raw Materials)
13. Review of Batch Quantities for Validation (Packing Materials)
14. HSE Requirements
15. Review of Process Parameters
16. Validation Procedure
17. Sampling Location
18. Documentation
19. Acceptance Criteria
20. Summary
21. Conclusion
Process Validation Protocol – Pharmaceutical Template PDF PPT XLS
PROCESS VALIDATION PROTOCOL -Pharmaceutical (Autoclave)
1. PRE-EXECUTION APPROVAL
Successful completion of this protocol will provide documented evidence that all key aspects of the Autoclave used in LARGE VOLUME PARENTRALS SECTION adheres to appropriate application criteria, comply with standard operating procedures, and meet current Good Manufacturing Practices (cGMP) requirements.
1.1 SIGNATORY LIST
The signature below indicates approval of this protocol and its attachments for execution.
(Name & Designation, Signature, Date, Prepared By, Checked and Reviewed By, Approved By are the rows and columns you need to fill in the signatory list)
|
Name & Designation |
Signature |
Date |
|
|
|
|
| PreparedBy |
|
|
|
|
|
|
|
| Checkedand Reviewed By |
|
|
|
|
|
|
|
| ApprovedBy |
|
|
|
1.2 Validation Team
All individuals participating in the execution of this protocol must fill out a row in the table below. with all the details like Name & Designation, Responsibility, Signature & Initial along with the Date of the process.
Prepare the protocol and coordinate the validation study. Generate amendments to the protocol as required
Microbiological validation of the sterilization process. document the microbiological aspects of the study
Protocol training of operators and provide the resources for validation study
3.0 INSTRUCTIONS
3.1. General Instruction
All performers and reviewers must complete qualification forms using the following guidelines:
· Complete all items on a form in full, except the optional comment’s section.
· Document any deviation from defined protocols and expected results. Owner approval of protocol deviations must be documented before final approval signatures can be obtained.
· Write additional comments on an addendum sheet when there is not enough space on a form to accommodate all comments. Use these three steps when adding an addendum sheet.
1. Number the page alphanumerically.
2. Initial and date additions.
3. Insert the addendum sheet behind the original page.
· Make all entries in permanent black or blue ball pen.
3.2 Correcting Entries
If you need to make corrections on a form, use the procedures described below:
3.2.1 Correcting Short Entries
To correct a short entry [such as a single word or test result] on a form:
1. Draw a diagonal line, bottom left to upper right, through the miss entered or incorrect information.
2. Write the correction to the upper right of the original entry.
3. Give brief explanation of change
4. Initial and date the change.
3.2.2 Correcting Long Entries
To correct a long entry or information block on a form:
1. Draw a diagonal line, bottom left to upper right, through the miss entered or incorrect information.
2. Write the correction on a separate addendum page.
3. Give brief explanation of change.
4. Initial and date the changes.
5. Number the page alphanumerically
6. Place the addendum page behind the original page.
3.3 Marking Elements That Are Not Applicable
Mark each element carefully according to the instruments below, so that it will be clear that the element is unnecessary and that you have not skipped or forgotten the element.
1. Draw a diagonal line, bottom left to upper right corner, through the element that is not required.
2. Write the letters NA [Not Applicable], your initials, and the date above the line. Include comments above the line or on the form to document the reason the element is not required.
3. Where NA is indicated as an option, select this field.
The performer and reviewer must sign and date all forms, as usual, even when part or all of the form is marked “NA”.
Note: All original entries must remain legible after any corrections have been made.
3.4 Caution
The following conditions require “re-qualification”;
· When a Instrument modification has been completed, it affects the installation qualification.
· When the software or firmware has been upgraded or changed
· When this Instrument is being removed from where it was originally installed.
3.5 Re-calibration / Re-certification Requirements
The following conditions require “re-calibration / re-certification;
· For a pre-determined period of time or use.
· After any minor service has been done or replacement of parts.
· When this Instrument is being removed from where it was originally installed.
4. RESPONSIBILITIES
4.1 Validation Team
· Prepare and approve the validation protocol.
· Provide training to the personnel regarding protocol execution.
· Assure complete adherence to the protocol during the execution
· Generate amendment to the validation protocol, as required.
· Document any deviations that occur during protocol execution.
· Document Operator SOP Training.
· Provide the resources required in executing the validation protocol.
4.2 PRODUCTION MANAGER
· Review the validation protocol and the final reports
4.3 QUALITY CONTROL/ASSURANCE MANAGER
· Approve the validation protocol and the final reports
5.0 Objectives:
To verify and establish that the Autoclave is working as per recommendations of the manufacturer.
6.0 Scope:
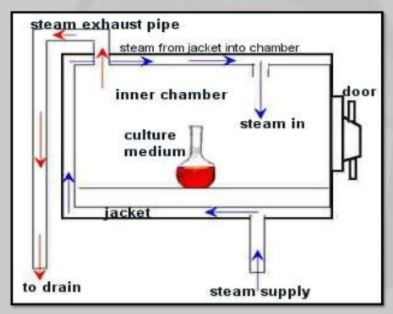
This validation protocol is applicable to the Autoclave intended to be used for steam sterilization in LARGE VOLUME PARENTRALS SECTION.
The protocol will be implemented under the following conditions
§ The validation of sterilization process using saturated steam as the steriliant
§ Prior to the production of a new sterilizer.
§ A change In the load design or weight that would result in a load that is more difficult to sterilize.
7.0 Equipment Identification
Qualification of utilities and equipment generally includes the following activities:
• Selecting utilities and equipment construction materials, operating principles, and performance characteristics based on whether they are appropriate for their specific uses.
• Verifying that utility systems and equipment are built and installed in compliance with the design specifications (e.g., built as designed with proper materials, capacity, and
functions, and properly connected and calibrated).
• Verifying that utility systems and equipment operate in accordance with the process requirements in all anticipated operating ranges.
Equipment Name
Autoclave
Time controller
¨
2
Pressure controller
¨
2
Temperature controller
¨
3
Pressure gauge
¨
4
Safety Valve
¨
5
Thermometer
¨
Completed By:__________________ Date:_____________
Reviewed By:___________________ Date:_____________
8.0 EQUIPMENT DESCRIPTION
The Autoclave intended to be used for steam sterilizations process. It has following specifications:-
S. No.
Parameter
Range
Readability
Check
01
Timer
0—60 min
1 min
¨
02
Pressure
0—60 Lb/inch²
2.0 Lb/inch²
¨
03
Temperature
0 –150°C
0.5°C
¨
8.1 LOAD IDENTIFICATION
Nature of load
1000ml bottles
Quantity of load
2000 Bottles
8.2 STERILIZATATION CYCLE PARAMETERS
Sterilization set point
106°C
Temperature range
106°C +0.5°C
Expose time
45 minutes
8.3 Equipment Used for PROCESS VALIDATION
Equipment
Calibration
Certificate No.
Issue Date
YES
NO
Recording potentiometer
¨
¨
___________
________
Thermocouples & lead wires
¨
¨
___________
________
Biological indicator i.e.
B. stereothermophyllus
¨
¨
___________
________
Completed By:__________________ Date:_____________
Reviewed By:___________________ Date:_____________
9.0 strerilizatation procedure:
§ Place six thermocouples in the load at the slow to heat points as determined
Previously by(Heat Distribution and Heat Penetration studies)
§ Place thermocouples exterior and near to (Penetration TC)and expose to chamber steam distribution TC)
§ Place BIs (Biological Indicators) at each of the slow to heat penetration location.
§ Load autoclave extend TC out of autoclave and attach to potentiometer
§ Position one TC by controller record sensor
§ Close autoclave door
§ Perform, function check of TC .replace if defective.
§ Replace autoclave sensor chart with a new one
§ Check to make sure that cycle parameters are set
§ Set potentiometer for a 3.0 Hours scan cycle.
§ Initiate sterilization cycle and potentiometer cycle at same time
§ Allow cycle to continue until it is completed. Collect all potentiometers, controls and computer control record and place with protocol.
§ Have computer graph results and calculate Fo value. After load has cooled, remove BIs and have tested
§ Incubate BIs in incubator at 55Cº for 48 hrs
10.0 ACCEPTANCE CRITERIA
1- BDS Strip
All four colors segment of the processed indicator are black. If all other critical process parameters such as temperature, pressure and sterilization are in accordance with cycle reference.
2- Bio-Indicator i.e. B. stereothermophyllus
No growth should be observed after incubation for 48 Hours.
10.1 Results
Temperature : 106°C
Pressure : 10 Lb/inch²
Sterilization Time : 30 minutes
1- Evaluation of the BDS strip.
S.#.
Position of Indicator strip
Stick BDS-test indicator strip on
Acceptance Criteria
Results
All four color segment of indicators strip are black
Yes
No
1
¨
¨
2
¨
¨
2- Evaluation of the Bio-indicator i.e. B. stereothermophyllus
S.#.
Position of
B. stereothermophyllus
Acceptance Criteria
Observation
No growth is observed after incubation for 48 Hours
Yes
No
1
Front/top
left
Fornt/bttm
center
Middle
/centleft
¨
¨
2
Middle/
bttmleft
Rare/top
center
Rare/bttm
left
¨
¨
3
Front/top
center
Front/bottm
center
Middle/cent
left
¨
¨
4
Middle/bttm
right
Rare/top
bottom
Rare/bottm
center
¨
¨
5
Front/top
right
Front/
bottmleft
Middle/
center
¨
¨
6
Middle/
Bttm/Cent
Rare/top
right
Rare/bttm
center
¨
¨
All acceptance criteria have been met. Verified By / Date
Yes ____________No _____________ _____________
If No or N/A, explain in Comments.
Comments:_____________________________________________________________
Completed By:__________________ Date:_____________
Reviewed By:________________ Date:_____________
11.0. Incidents/Deviations
To document any discrepancy or variations noted during the execution of the Process Validation Protocol. Any action to be taken to resolve an outstanding issue is to be identified within the incident report.
INCIDENT #
DESCRIPTION OF INCIDENT
RECORDED BY
DATE
COMMENTS:
____________________________________________________________
____________________________________________________________
12.0 FINAL COMMENTS ABOUT PROCESS VALIDATION
________________________________________________________________
________________________________________________________________
________________________________________________________________
________________________________________________________________
13.0 SIGNATURE IDENTIFICATION SHEET
This sheet is a record of each individual who signs or initials any page included in this protocol or in the attached document. Each person shall be identified by typed or printed name.
Name Signature and Initials Company
__________________ _________________________ _____________________
__________________ _________________________ _____________________
__________________ _________________________ _____________________
__________________ _________________________ _____________________
__________________ _________________________ _____________________
__________________ ________________________ _____________________
__________________ ________________________ _____________________
__________________ ________________________ _____________________
__________________ _________________________ _____________________
FINAL APPROVAL OF QUALIFICATION
This document certifies that the process of Autoclavation has been validated as specified and complies with Standard Operating Procedures, and satisfies the requirements for cGMPs.
Name & Designation
Signature
Date
Prepared By
Tahir Ibrahim
Quality Assurance Executive
checked and Reviewed By
Abdul Hafeez
Production manager
Approved By
Tajjamal A Qurashi
Manager Quality Control
PROTOCOL TRAINING
Training Session Date : ____________________
Instructor : ____________________
Protocol Reference : ____________________
Name
Title
Signature
Date
PROCESS VALIDATION PROTOCOL -Pharmaceutical Template PDF PPT XLS
In conclusion, there is far to think about about your Computer System Validation system last to a strong inspection. Make every effort to have a system stock list put in place, the appropriate SOPs in place, and additionally to check the critical techniques and their documentation just before a powerful FDA inspection. Again, simply because the FDA can be inspecting the institution for other factors, doesn’t discount the potential the couple need to audit your pc System Validation School. As mentioned, so many of our businesses respective company procedures are carried out by way of electronic systems in this young age of technologies. Therefore, it could be useful to evaluate the Computer Validation Program whether you foresee a strong inspection or otherwise not. Having a powerful efficient Computer System Validation System put in place will help ensure the stability of the electronic documents, allocate resources better and subsequently can yield long run cost discounts to the company.
more information
DOCUMENTATION
Documentation at each stage of the process validation lifecycle is essential for effective communication in complex, lengthy, and multidisciplinary projects. Documentation is important
so that knowledge gained about a product and process is accessible and comprehensible to others involved in each stage of the lifecycle. Information transparency and accessibility are
fundamental tenets of the scientific method. They are also essential to enabling organizational units responsible and accountable for the process to make informed, science-based decisions that
ultimately support the release of a product to commerce.
The degree and type of documentation required by CGMP vary during the validation lifecycle. Documentation requirements are greatest during Stage 2, process qualification, and Stage 3,
continued process verification. Studies during these stages must conform to CGMPs and must be approved by the quality unit in accordance with the regulations .
Viral and impurity clearance studies, even when performed at small scale, also require quality
unit oversight.
CGMP documents for commercial manufacturing (i.e., the initial commercial master batch production and control record and supporting procedures) are key outputs of Stage 1,
process design. We recommend that firms diagram the process flow for the full-scale process.
Process flow diagrams should describe each unit operation, its placement in the overall process, monitoring and control points, and the component, as well as other processing material inputs
(e.g., processing aids) and expected outputs (i.e., in-process materials and finished product). It is also useful to generate and preserve process flow diagrams of the various scales as the process
design progresses to facilitate comparison and decision making about their comparability.
In conclusion, there is far to think about about your Computer System Validation system last to a strong inspection just before a powerful FDA inspection. Again, simply because the FDA can be inspecting the institution for other factors, doesn’t discount the potential the couple need to audit your pc System Validation School. As mentioned, so many of our businesses respective company procedures are carried out by way of electronic systems in this young age of technologies. Therefore, it could be useful to evaluate the Computer Validation Program whether you foresee a strong inspection or otherwise not.
GLOSSARY OF TERMS
2.1 List of Abbreviation
CGMP Current Good Manufacturing Practices
FDA Food and Drug Administration
GAMP Good Automated Manufacturing Practice
GMP Good Manufacturing Practice
IQ Installation Qualification
OQ Operation Qualification
2.2 Definitions
Acceptance Criteria Agreed standards or ranges, which must be achieved.
Critical component A component within a system where the operation, contact, data, control, alarm, or failure may have a direct impact on the quality of the product.
Critical Instrument Any instrument that directly affects product safety, purity, or efficacy.
Direct Impact System An engineering system that may have a direct impact on product quality.
Factor Acceptance Test Documenting the performance characteristics of equipment prior to shipment to site.
Impact Assessment The process of evaluating the impact of the operating, controlling alarming and failure conditions of a system on the quality of a product.
Indirect Impact System An engineering system considered not having a direct impact on product quality.
Installation Qualification Documenting the process equipment and ancillary system are constructed and installed according to pre-determined specifications and functional requirements.
No Impact System This is a system that will not have any impact, either directly or indirectly, on product quality. These systems are designed and commissioned following Good engineering Practice only.
Non-critical Component A component within a system where the operation, contact, alarm or failure may have an indirect impact or no impact on the quality of product.
Operating Limits The minimum and /or maximum values that will ensure that product and safety requirements are met.
Operational Qualification Establishing confidence that process equipment and ancillary systems are capable of consistently operating within established limits and tolerances.
Performance Qualification The documented verification that al aspects of a facility, utility or equipment that can affect product quality perform as intended meeting pre-determined acceptance criteria.
Performance Testing The process by which the performance of interdependent system is demonstrated as within the required tolerances, the output of interdependent system is demonstrated as delivering the required duty or capacity, the interdependent functions of system are interdependent to be as specified and appropriate.
Piping and Instrumentation
Diagrams Primary source of design information for utility systems and process equipment. They are used to depict the process flow, equipment configuration, process parameters, instrumentation, and materials of construction. They also are used to perform overall material and energy balances and pressure balances.
Capability of a process: Ability of a process to produce a product that will fulfill the requirements of that product. The concept of process capability can also be defined in statistical terms. (ISO 9000:2005)
Commercial manufacturing process: The manufacturing process resulting in commercial product (i.e., drug that is marketed, distributed, and sold or intended to be sold). For the purposes of this guidance, the term commercial manufacturing process does not include clinical trial or treatment IND material.
Concurrent release: Releasing for distribution a lot of finished product, manufactured following a qualification protocol, that meets the lot release criteria established in the protocol, but before the entire study protocol has been executed.
Continued process verification: Assuring that during routine production the process remains in a state of control. Performance indicators: Measurable values used to quantify quality objectives to reflect the performance of an organization, process or system, also known as performance metrics in some regions. (ICH Q10)
Process design: Defining the commercial manufacturing process based on knowledge gained through development and scale-up activities.
Process qualification: Confirming that the manufacturing process as designed is capable of reproducible commercial manufacturing.
Process validation: The collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality products.
Quality: The degree to which a set of inherent properties of a product, system, or process fulfils requirements. (ICH Q9)
State of control: A condition in which the set of controls consistently provides assurance of continued process performance and product quality. (ICH Q10)

![[PPT PDF] Pharmaceutical Water System Design Validation -SAMPLING CONSIDERATIONS](https://pharmawiki.in/wp-content/uploads/2017/11/PPT-PDF-Pharmaceutical-Water-System-Design-Validation-SAMPLING-CONSIDERATIONS.jpg)

![[PDF PPT] Biotechnology Plant Design - Biotech Laboratory DESIGN CONSTRUCTION & VALIDATION OF GMP](https://pharmawiki.in/wp-content/uploads/2017/04/Biotechnology-Plant-Design-1.jpg)