Sickle cell disease is an important genetic cause of hemolytic anemia, a form of anemia due to increased erythrocyte destruction, instead of the reduced mature erythrocyte production seen with iron, folic acid, and vitamin B 12 deficiency. Patients with sickle cell disease are homozygous for the aberrant β-hemoglobin S (HbS) allele or heterozygous for HbS and a second mutated β-hemoglobin gene such as hemoglobin C ( HbC ) or β-thalassemia. Sickle cell disease has an increased prevalence in individuals of African descent because the heterozygous trait confers resistance to malaria. In the majority of patients with sickle cell disease, anemia is not the major problem; the anemia is generally well compensated even though such individuals have a chronically low hematocrit (20–30%), a low serum hemoglobin level (7–10 g/dL), and an elevated reticulocyte count. Instead, the primary problem is that deoxygenated HbS chains form polymeric structures that dramatically change erythrocyte shape, reduce deformability, and elicit membrane permeability changes that further promote hemoglobin polymerization. Abnormal erythrocytes aggregate in the microvasculature—where oxygen tension is low and hemoglobin is deoxygenated—and cause veno-occlusive damage. The clinical manifestations of sickle cell disease reflect organ damage by veno-occlusive events. In the musculoskeletal system, this results in characteristic, extremely painful bone and joint pain. In the cerebral vascular system, it causes ischemic stroke. Damage to the spleen increases the risk of infection, particularly by encapsulated bacteria such as Streptococcus pneumoniae . In the pulmonary system, there is an increased risk of infection and, in adults, an increase in embolism and pulmonary hypertension. Supportive treatment includes analgesics, antibiotics, pneumococcal vaccination, and blood transfusions. In addition, the cancer chemotherapeutic drug hydroxyurea (hydroxycarbamide) reduces veno-occlusive events. It is approved in the United States for treatment of adults with recurrent sickle cell crises and approved in Europe in adults and children with recurrent vaso-occlusive events. As an anticancer drug used in the treatment of chronic and acute myelogenous leukemia, hydroxyurea inhibits ribonucleotide reductase and thereby depletes deoxynucleoside triphosphate and arrests cells in the S phase of the cell cycle (see Chapter 54 ). In the treatment of sickle cell disease, hydroxyurea acts through poorly defined pathways to increase the production of fetal hemoglobin γ (HbF), which interferes with the polymerization of HbS. Clinical trials have shown that hydroxyurea decreases painful crises in adults and children with severe sickle cell disease. Its adverse effects include hematopoietic depression, gastrointestinal effects, and teratogenicity in pregnant women.
World SICKLE CELL Day – DATE JUNE 19TH
SICKLE CELL ANEMIA:
The term sickle cell disease encompasses a variety of hemoglobinopathies, including sickle cell anemia, sickle Hb C (SC) disease, & sickle cell thalassemia. Although the clinical presentations of all are often similar, the manifestations of sickle cell are more severe & so mainly considered.
PATHOPHYSIOLOGY:
Hb is distinguished as Hb A1, HbA2, Hb C, Hb F & Hb S of which Hb A1, Hb A2 & Hb F are normal. Hb A1 – a tetramer consist 2 pairs of globin chains α & β. Substitution of valine for glutamic acid in both the β chains. Each parent contributes a single β chain gene, the heterozygous genotype AS is also possible & is expressed as the sickle cell trait phenotype.
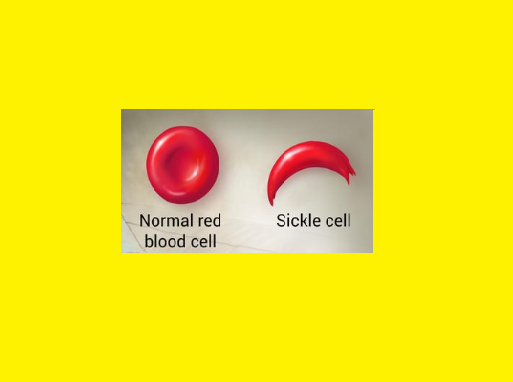
Deoxygenation in capillaries induces rapid polymerization of the sickling Hb, Hb S & results in formation of helical strands of parallel fibres.
The elongated, crescent shaped cells characteristic of sickle cell anemia are so produced. The affected erythrocytes are rigid & unable to pass through the microvasculature. Vasooclusion with subsequent painful ischemia & chronic organ damage.
Sickling is reversible upon reexposure to oxygen, however repeated sickling episodes eventually damage the cell membrane.
The rate of Hb polymerization depends on its concentration in the erythrocyte. The co polymerization of Hb S with Hb F inhibits further polymer growth ; intracellular Hb F concentrations are inversely correlated with severity of disease.
Sicke cell anemia symptoms & clinical presentations:
– Impaired growth & development – Increased risk of infection viz meningitis, pneumonia, septicemia Hematologic – Hemolytic anemia – Aplastic crises – Splenic sequestration crises Vasoocclusive : 1. Cardiovascular: – Cardiac enlargement – Priapism – Renal insufficiency Painful crises – Systolic murmur 7. Pulmonary: 4. Neurologic : – Acute chest syndrome 2. GI: – Autosplenecetomy – Gallstones / cholecystitis – Cerebral thrombosis – Intracerebral hemorrhage – Seizures – Chronic obstructive disease – Infarction – Hepatic insufficiency – Intrahepatic cholelithiais – Subarachnoid haemorrhage 8. Skin & skeletal : – Arthropathy 5. Ocular: – Aseptic necrosis 3. Genitourinary: – Hematuria – Impotence – Retinopathy – Secondary glaucoma- Leg ulcers
Diagnosis of Sickle cell anemia:
Hb electrophoresis types & proportion of Hb present.
Is rapid & inexpensive screening test
It establishes the patients genotype.
If both parents have the AS genotype there is a 1 in 4 chance that their child will have homozygous SS disease.
Prenatal diagnosis also possible
TREATMENT:
1. Management of major complications:
a. Anemia
Blood transfusions
Folate supplementations
b. Infection
Cefuroxime for Pneumonia & erythromycin & azithromycin for Mycoplasma pneumonia treatment. Prophylactic penicillin for pneumococcal septicemias.
Ampicillin & cephalosporins for salmonella infections.
c. Painful crisis
Vigorous hydration is initiated & oxygen administered if hypoxia is present.
Ketorolac is given if codeine or oxycodones singly or in combination with acetaminophen are not effective.
2. Management of the sickle cell disease:
a. Transfusion therapy
b. Pharmacologic management : clotrimazole. Pentoxiphylline, antineoplastics, hydroxyurea.
c. Bone marrow transplantation