Pharmacodynamics Definition:
Pharmacodynamics the branch of pharmacology concerned with the effects of drugs and the mechanism of their action.
“Pharmacodynamics involves how the drugs act on target cells to alter cellular function.”
A. Receptor and non-receptor mechanisms: Most of the drugs act by interacting with a cellular component called receptor. Some drugs act through simple physical or chemical reactions without interacting with any receptor.
• Receptors are protein molecules present either on the cell surface or with in the cell e.g. adrenergic receptors, cholinoceptors, insulin receptors, etc.
• The endogenous neurotransmitters, hormones, autacoids and most of the drugs produce their effects by binding with their specific receptors.
• Aluminium hydroxide and magnesium trisilicate, which are used in the treatment of peptic ulcer disease act by non-receptor mechanism by neutralizing the gastric acid.
Pharmacodynamics Basics:
Many drugs are similar to or have similar chemical groups to the naturally occurring chemical and have the ability to bind onto a receptor where one of two things can happen- either the receptor will respond or it will be blocked.
A drug, which is able to fit onto a receptor, is said to have affinity for that receptor. Efficacy is the ability of a drug to produce an effect at a receptor. An agonist has both an affinity and efficacy whereas antagonist has affinity but not efficacy or intrinsic activity.
When a drug is able to stimulate a receptor, it is known as an agonist and therefore mimics the endogenous transmitter.
When the drug blocks a receptor, it is known as antagonist and therefore blocks the action of the endogenous transmitter (i.e. it will prevent the natural chemical from acting on the receptor).
However, as most drug binding is reversible, there will be competition between the drug and the natural stimulus to the receptor.
Pharmacodynamics Basic Notes – PDF PPT – ATROPINE FUROSIMIDE HEPARIN BASTI VAMANA
The forces that attract the drug to its receptor are termed chemical bonds and they are
(a)hydrogen bond
(b) ionic bond
(c) covalent bond
(d) Vander waals force.
Covalent bond is the strongest bond and the drug-receptor complex is usually irreversible.
K1 K3
DR Biological effect
D+R K2
Where D = Drug, R= receptor DR= Drug receptor complex (affinity)
K1 = association constant
K2 = dissociation constant
K3 = intrinsic activity
When first messengers like neurotransmitters, hormones, autacoids and most of drugs bind with their specific receptors, the drug receptor complex is formed which subsequently causes the synthesis and release of another intracellular regulatory molecule termed as second messengers e.g. cyclic AMP, calcium, cyclic GMP, inositol triphosphate (IP3), diacylglycerol and calmodulin which in turn produce subcellular or molecular mechanism of drug action.
B. Site of drug action:
– A drug may act:
(i) Extracellularly e.g: osmotic diuretics, plasma expanders.
(ii) On the cell surface e.g.: digitalis, penicillin, catecholamines
(iii) Inside the cell e.g.: anti-cancer drugs, steroid hormones.
C. Dose Response relationship
The exact relationship between the dose and the response depends on the biological object under observation and the drug employed.
When a logarithm of dose as abscissa and responses as ordinate are constructed graphically, the “S” shaped or sigmoid type curve is obtained.
The lowest concentration of a drug that elicits a response is minimal dose, and the largest concentration after which further increase in concentration will not change the response is the maximal dose.
1. Graded dose effect: As the dose administered to a single subject or tissue increases, the pharmacological response also increases in graded fashion up to ceiling effect.
– It is used for characterization of the action of drugs. The concentration that is required to produce 50 % of the maximum effect is termed as EC50 or ED50.50
2. Quantal dose effect: It is all or none response, the sensitive objects give response to small doses of a drug while some will be resistant and need very large doses. The quantal dose effect curve is often characterized by stating the median effective dose and the median lethal dose.
Median lethal dose or LD50: This is the dose (mg/kg), which would be expected to kill one half of a population of the same species and strain.
Median effective dose or ED50: This is the dose (mg/kg), which produces a desired response in 50 per cent of test population.
Therapeutic index: It is an approximate assessment of the safety of the drug. It is the ratio of the median lethal dose and the median effective dose. Also called as therapeutic window or safety.
The larger the therapeutic index, the safer is the drug. Penicillin has a very high therapeutic index, while it is much smaller for the digitalis preparation.
D. Structural activity relationship
The activity of a drug is intimately related to its chemical structure. Knowledge about the chemical structure of a drug is useful for:
(i) Synthesis of new compounds with more specific actions and fewer adverse reactions
(ii) Synthesis of competitive antagonist and
(iii) Understanding the mechanism of drug action.
Slight modification of structure of the compound can change the effect completely.
pharmacodynamics example, pharmacodynamics basics, pharmacodynamics pdf, pharmacodynamics ppt, pharmacodynamics vs pharmacokinetics, pharmacodynamics definition nursing, pharmacodynamics slideshare, pharmacodynamics what the drug does to the body, pharmacodynamics of atropine, pharmacodynamics of furosimide, pharmacodynamics ofheparin, pharmacodynamics of salbutamol pharmacodynamics of vamana, pharmacodynamics of basti, pharmacodynamics of paracetamol, pharmacodynamics of phenytoin, pharmacodynamics of aspirin, pharmacodynamics of pantaprazole, pharmacodynamics example, pharmacodynamics basics, pharmacodynamics pdf, pharmacodynamics ppt, pharmacodynamics vs pharmacokinetics
Pharmacodynamics Examples:

Pharmacodynamics of atropine:
Atropine, a naturally occurring belladonna alkaloid, is a racemic mixture of equal parts of d- and l-hyoscyamine, whose activity is due almost entirely to the levo isomer of the drug. Atropine is commonly classified as an anticholinergic or antiparasympathetic (parasympatholytic) drug. More precisely, however, it is termed an antimuscarinic agent since it antagonizes the muscarine-like actions of acetylcholine and other choline esters. Adequate doses of atropine abolish various types of reflex vagal cardiac slowing or asystole. The drug also prevents or abolishes bradycardia or asystole produced by injection of choline esters, anticholinesterase agents or other parasympathomimetic drugs, and cardiac arrest produced by stimulation of the vagus. Atropine may also lessen the degree of partial heart block when vagal activity is an etiologic factor. Atropine in clinical doses counteracts the peripheral dilatation and abrupt decrease in blood pressure produced by choline esters. However, when given by itself, atropine does not exert a striking or uniform effect on blood vessels or blood pressure.
Pharmacodynamics of Furosemide
Furosemide, a sulfonamide-type loop diuretic structurally related to bumetanide, is used to manage hypertension and edema associated with congestive heart failure, cirrhosis, and renal disease, including the nephrotic syndrome.
Furosemide, a loop diuretic, inhibits water reabsorption in the nephron by blocking the sodium-potassium-chloride cotransporter (NKCC2) in the thick ascending limb of the loop of Henle. This is achieved through competitive inhibition at the chloride binding site on the cotransporter, thus preventing the transport of sodium from the lumen of the loop of Henle into the basolateral interstitium. Consequently, the lumen becomes more hypertonic while the interstitium becomes less hypertonic, which in turn diminishes the osmotic gradient for water reabsorption throughout the nephron. Because the thick ascending limb is responsible for 25% of sodium reabsorption in the nephron, furosemide is a very potent diuretic.
Pharmacodynamics of Heparin
Unfractionated heparin is a highly acidic mucopolysaccharide formed of equal parts of sulfated D-glucosamine and D-glucuronic acid with sulfaminic bridges. The molecular weight ranges from 3000 to 30,000 daltons. Heparin is obtained from liver, lung, mast cells, and other cells of vertebrates. Heparin is a well-known and commonly used anticoagulant which has antithrombotic properties. Heparin inhibits reactions that lead to the clotting of blood and the formation of fibrin clots both in vitro and in vivo. Small amounts of heparin in combination with antithrombin III, a heparin cofactor,) can inhibit thrombosis by inactivating Factor Xa and thrombin. Once active thrombosis has developed, larger amounts of heparin can inhibit further coagulation by inactivating thrombin and preventing the conversion of fibrinogen to fibrin. Heparin also prevents the formation of a stable fibrin clot by inhibiting the activation of the fibrin stabilizing factor. Heparin prolongs several coagulation tests. Of all the coagulation tests, activated partial prothrombin time (aPTT) is the most clinically important value.
Mechanism of action
Under normal circumstances, antithrombin III (ATIII) inactivates thrombin (factor IIa) and factor Xa. This process occurs at a slow rate. Administered heparin binds reversibly to ATIII and leads to almost instantaneous inactivation of factors IIa and Xa The heparin-ATIII complex can also inactivate factors IX, XI, XII and plasmin. The mechanism of action of heparin is ATIII-dependent. It acts mainly by accelerating the rate of the neutralization of certain activated coagulation factors by antithrombin, but other mechanisms may also be involved. The antithrombotic effect of heparin is well correlated to the inhibition of factor Xa. Heparin is not a thrombolytic or fibrinolytic. It prevents progression of existing clots by inhibiting further clotting. The lysis of existing clots relies on endogenous thrombolytics.
Pharmacodynamics of paracetamol
Pharmacodynamics of Acetaminophen
Acetaminophen (USAN) or Paracetamol (INN) is a widely used analgesic and antipyretic drug that is used for the relief of fever, headaches, and other minor aches and pains. It is a major ingredient in numerous cold and flu medications and many prescription analgesics. It is extremely safe in standard doses, but because of its wide availability, deliberate or accidental overdoses are not uncommon. Acetaminophen, unlike other common analgesics such as aspirin and ibuprofen, has no anti-inflammatory properties or effects on platelet function, and it is not a member of the class of drugs known as non-steroidal anti-inflammatory drugs or NSAIDs. At therapeutic doses acetaminophen does not irritate the lining of the stomach nor affect blood coagulation, kidney function, or the fetal ductus arteriosus (as NSAIDs can). Like NSAIDs and unlike opioid analgesics, acetaminophen does not cause euphoria or alter mood in any way. Acetaminophen and NSAIDs have the benefit of being completely free of problems with addiction, dependence, tolerance and withdrawal. Acetaminophen is used on its own or in combination with pseudoephedrine, dextromethorphan, chlorpheniramine, diphenhydramine, doxylamine, codeine, hydrocodone, or oxycodone.
Mechanism of action:
Acetaminophen is thought to act primarily in the CNS, increasing the pain threshold by inhibiting both isoforms of cyclooxygenase, COX-1, COX-2, and COX-3 enzymes involved in prostaglandin (PG) synthesis. Unlike NSAIDs, acetaminophen does not inhibit cyclooxygenase in peripheral tissues and, thus, has no peripheral anti-inflammatory affects. While aspirin acts as an irreversible inhibitor of COX and directly blocks the enzyme’s active site, studies have found that acetaminophen indirectly blocks COX, and that this blockade is ineffective in the presence of peroxides. This might explain why acetaminophen is effective in the central nervous system and in endothelial cells but not in platelets and immune cells which have high levels of peroxides. Studies also report data suggesting that acetaminophen selectively blocks a variant of the COX enzyme that is different from the known variants COX-1 and COX-2. This enzyme is now referred to as COX-3. Its exact mechanism of action is still poorly understood, but future research may provide further insight into how it works. The antipyretic properties of acetaminophen are likely due to direct effects on the heat-regulating centres of the hypothalamus resulting in peripheral vasodilation, sweating and hence heat dissipation.
Pharmacodynamics of salbutamol
Salbutamol (INN) or albuterol (USAN), a moderately selective beta(2)-receptor agonist similar in structure to terbutaline, is widely used as a bronchodilator to manage asthma and other chronic obstructive airway diseases. The R-isomer, levalbuterol, is responsible for bronchodilation while the S-isomer increases bronchial reactivity. The R-enantiomer is sold in its pure form as Levalbuterol. The manufacturer of levalbuterol, Sepracor, has implied (although not directly claimed) that the presence of only the R-enantiomer produces fewer side-effects.
Mechanism of action:
Salbutamol is a beta(2)-adrenergic agonist and thus it stimulates beta(2)-adrenergic receptors. Binding of albuterol to beta(2)-receptors in the lungs results in relaxation of bronchial smooth muscles. It is believed that salbutamol increases cAMP production by activating adenylate cyclase, and the actions of salbutamol are mediated by cAMP. Increased intracellular cyclic AMP increases the activity of cAMP-dependent protein kinase A, which inhibits the phosphorylation of myosin and lowers intracellular calcium concentrations. A lowered intracellular calcium concentration leads to a smooth muscle relaxation and bronchodilation. In addition to bronchodilation, salbutamol inhibits the release of bronchoconstricting agents from mast cells, inhibits microvascular leakage, and enhances mucociliary clearance.
Pharmacodynamics of vamana
The overall Pharmacodynamic of Vamanopaga dasemāni drugs is based on guna concept. Most of the drugs (90%) are having property of Laghu and Ruksa guna. These are based on Vāyu, Agni and Ākasa mahābhaūtik (one of the five elements of the universe) composition. Ācarya Caraka has mentioned only the role of gunas in the Pharmacodynamic of Vamana karma (Bhadanta Nāgārjunā, Rasavaisesika, 2010). In fact guna is the thing
which represents a drug. So, the selection of a drug should be on the basis of gunas for Vamana karma.
Ācarya has mentioned predominance of Vāyu and Agni mahābhūta drugs for Vamana karma. Rasas (taste) of vamana dravyas are chiefly katu and kasāya rasa which are composition of the same mahābhūtas. Most of
drugs are katu Vipāka having similar bhaūtic constitution. Other drugs are supportive to the therapy or to avoid complications during Vamana karma. As an example; honey which is mentioned in Vamanopaga dasemāni is added
to Vamana kalpa (prepared medicine) for increasing the palatability and giving soothing effect. Āyurveda says it is a good kapha chedaka (expectorant), helps in better expulsion of malarūpī kapha by vamana karma. Likewise Saindhava (salt) should be added to Vamana kalpa for Vilāyana (Agnivesa, Caraka Samhita, 2001) (liquefying)
of sticky Kaphadosa in channels. Effect of both the drugs is to help in a comfortable and irritation less procedure. added to Vamana kalpa for Vilāyana (Agnivesa, Caraka Samhita, 2001) (liquefying) of sticky Kaphadosa in channels. Effect of both the drugs is to help in a comfortable and irritation less procedure.
Pharmacodynamics of basti
Basti is chief Panchakama procedure used in Ayurveda. The pharmacodynamics of systemic effect of Basti may be understood through absorption mechanism, concept of system biology, neural stimulation mechanism, and excretory mechanism. As Basti is homogenous emulsion mixture of Honey, Saindhava,Sneha Dravya, Kalka, and decoction of crude drugs and Prakshepa Dravya, which is given through rectum, is absorbed, hence Basti is used as route of drug administration. Through rectal route large quantity of drugs can be delivered for systemic circulation and act accordingly. Concept of system biology opines that a change at cellular level of a system can bring changes in tissue, organ and system and in another system consequently & finally in whole body. As per recent advancement intestine not only is highly vascular but also highly innervated organ which forms ‘Enteric Nervous System’ (ENS).ENS may works in synergism with Central Nervous System of body. The cleansing action of Basti is related with the facilitation of excretion of morbid substances responsible for the disease process into the colon, from where it is evacuated.
Basti being the most widely used and highly effective treatment modality in the Ayurveda, it is the prime subject of interest for modern scientific community. With this background the basic question which comes forward regarding Basti is, “do active principles of drugs used in Basti get absorbed in systemic circulation. Triphaladi decoction Basti containing biomarker gallic acid and after Basti they traced it in the circulation. The rectum has rich blood and lymph supply and drugs can cross the rectal mucosa like other lipid membrane. Thus unionised and lipid soluble
substances are readily absorbed from the rectal mucosa. Small quantity of short chain fatty acid fatty acids, such as those from butterfat are absorbed directly into portal blood rather than being converted into triglycerides. This is because short chain fatty acids are more water soluble and allow direct diffusion from the epithelial cells into
capillary blood of villi. However decoction Basti gets a very little time maximum 48 minutes to absorb from colon and rectum how so ever these areas have very large surface area and highly vascular needed for absorption. Retention time for Anuvashana Basti is relatively more so probability of absorption also increases. Anuvasana Basti
after reaching in the rectum and colon causes secretion of bile from gall bladder which leads to the formation of conjugate micelles which is absorbed through passive diffusion. Especially short chain fatty acid present in Sneha of
Anuvasana Basti may absorb from colon and large intestine part of gastrointestinal tract and break the pathology of disease. In Basti Karma, a homogenous emulsion
2) By System Biology Concept of Honey, Saindhava, Sneha Dravya, Kalka, and decoction mixed in remarkable combination after proper churning (break the large and middle chain fatty acid into small chain fatty acids) is given which facilitates absorption better then a single drug per rectum. In Ayurveda classics, various Basti Dravya are
mentioned in diverse proportion in different diseases, it again confirms pharmacodynamics of Basti through absorption mechanism
Pharmacodynamics of phenytoin
Phenytoin is an antiepileptic drug which can be useful in the treatment of epilepsy. The primary site of action appears to be the motor cortex where spread of seizure activity is inhibited. Phenytoin reduces the maximal activity of brain stem centers responsible for the tonic phase of tonic-clonic (grand mal) seizures. Phenytoin acts to dampen the unwanted, runaway brain activity seen in seizure by reducing electrical conductance among brain cells. It lacks the sedation effects associated with phenobarbital. There are some indications that phenytoin has other effects, including anxiety control and mood stabilization, although it has never been approved for those purposes by the FDA. Phenytoin is primarily metabolized by CYP2C9.
Mechanism of action
Phenytoin acts on sodium channels on the neuronal cell membrane, limiting the spread of seizure activity and reducing seizure propagation. By promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of post-tetanic potentiation at synapses. Loss of post-tetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas.
Pharmacodynamics of Aspirin
Acetylsalicylic acid is an analgesic, antipyretic, antirheumatic, and anti-inflammatory agent. Acetylsalicylic acid’s mode of action as an antiinflammatory and antirheumatic agent may be due to inhibition of synthesis and release of prostaglandins. Acetylsalicylic acid appears to produce analgesia by virtue of both a peripheral and CNS effect. Peripherally, acetylsalicylic acid acts by inhibiting the synthesis and release of prostaglandins. Acting centrally, it would appear to produce analgesia at a hypothalamic site in the brain, although the mode of action is not known. Acetylsalicylic acid also acts on the hypothalamus to produce antipyresis; heat dissipation is increased as a result of vasodilation and increased peripheral blood flow. Acetylsalicylic acid’s antipyretic activity may also be related to inhibition of synthesis and release of prostaglandins.
Mechanism of action:
The analgesic, antipyretic, and anti-inflammatory effects of acetylsalicylic acid are due to actions by both the acetyl and the salicylate portions of the intact molecule as well as by the active salicylate metabolite. Acetylsalicylic acid directly and irreversibly inhibits the activity of both types of cyclooxygenase (COX-1 and COX-2) to decrease the formation of precursors of prostaglandins and thromboxanes from arachidonic acid. This makes acetylsalicylic acid different from other NSAIDS (such as diclofenac and ibuprofen) which are reversible inhibitors. Salicylate may competitively inhibit prostaglandin formation. Acetylsalicylic acid’s antirheumatic (nonsteroidal anti-inflammatory) actions are a result of its analgesic and anti-inflammatory mechanisms; the therapeutic effects are not due to pituitary-adrenal stimulation. The platelet aggregation-inhibiting effect of acetylsalicylic acid specifically involves the compound’s ability to act as an acetyl donor to cyclooxygenase; the nonacetylated salicylates have no clinically significant effect on platelet aggregation. Irreversible acetylation renders cyclooxygenase inactive, thereby preventing the formation of the aggregating agent thromboxane A2 in platelets. Since platelets lack the ability to synthesize new proteins, the effects persist for the life of the exposed platelets (7-10 days). Acetylsalicylic acid may also inhibit production of the platelet aggregation inhibitor, prostacyclin (prostaglandin I2), by blood vessel endothelial cells; however, inhibition prostacyclin production is not permanent as endothelial cells can produce more cyclooxygenase to replace the non-functional enzyme.
Pharmacodynamics of pantaprazole
Pantoprazole is a substituted benzimidazole indicated for the short-term treatment (up to 16 weeks) in the healing and symptomatic relief of erosive esophagitis. Pantoprazole is a proton pump inhibitor (PPI) that suppresses the final step in gastric acid production.
Mechanism of action:
Pantoprazole is a proton pump inhibitor (PPI) that suppresses the final step in gastric acid production by forming a covalent bond to two sites of the (H+,K+ )- ATPase enzyme system at the secretory surface of the gastric parietal cell. This effect is dose- related and leads to inhibition of both basal and stimulated gastric acid secretion irrespective of the stimulus.







 , per unit volume of total moist air,
, per unit volume of total moist air, 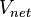 , can be expressed as follows:
, can be expressed as follows:
 in the mixture to the saturated vapor pressure of water
in the mixture to the saturated vapor pressure of water  at a prescribed temperature.
at a prescribed temperature.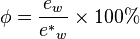
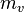 , per unit mass of dry air
, per unit mass of dry air  .
.
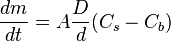
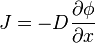
 is the “diffusion flux” [(amount of substance) per unit area per unit time], example
is the “diffusion flux” [(amount of substance) per unit area per unit time], example  .
. 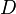 is the diffusion coefficient or diffusivity in dimensions of [length2 time−1], example
is the diffusion coefficient or diffusivity in dimensions of [length2 time−1], example 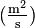
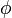 (for ideal mixtures) is the concentration in dimensions of [(amount of substance) length−3], example
(for ideal mixtures) is the concentration in dimensions of [(amount of substance) length−3], example 
 is the position [length], example
is the position [length], example 


 is time [s]
is time [s]
