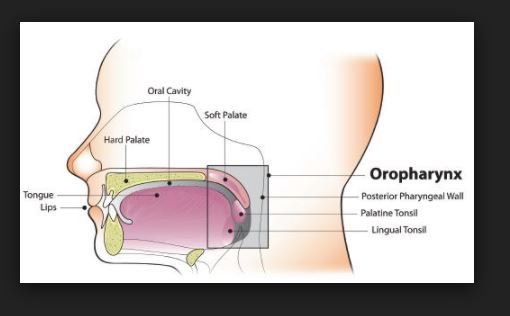
Anatomy and Physiology Of human respiratory system is a complicated organ system of very close structure– function relationships. The system consisted of two regions: the conducting airway and the respiratory region. The airway is further divided into many folds: nasal cavity and the associated sinuses, and the nasopharynx, oropharynx, larynx, trachea, bronchi, and bronchioles. The respiratory region consists of respiratory bronchioles, alveolar ducts, and alveolar sacs.
ANATOMY AND PHYSIOLOGY OF HUMAN RESPIRATORY TRACT:
The respiratory system works with the circulatory system to deliver oxygen from the lungs to the cells and remove carbon dioxide, and return it to the lungs to be exhaled. The exchange of oxygen and carbon dioxide between the air, blood and body tissues is known as respiration. Healthy lungs take in about 1 pint of air about 12–15 times each minute. All of the blood in the body is passed through the lungs every minute. The respiratory tract is divided into two main parts: the upper respiratory tract, consisting of the nose, nasal cavity and the pharynx; and the lower respiratory tract consisting of the larynx, trachea, bronchi and the lungs The trachea, which begins at the edge of the larynx, divides into two bronchi and continues into the lungs. The trachea allows air to pass from the larynx to the bronchi and then to the lungs. The bronchi divide into smaller bronchioles which branch in the lungs forming passageways for air. The terminal parts of the bronchi are the alveoli. The alveoli are the functional units of the lungs and they form the site of gaseous exchange
The blood barrier between the alveolar space and the pulmonary capillaries is very thin to allow for rapid gas exchange. During inspiration, oxygen diffuses through the alveoli walls and the interstitial space, into the blood. Carbon dioxide diffuses in the opposite direction during exhalation. Alveoli are small and there are approximately 300 million of them in each lung. Although alveoli are tiny structures, they have a very large surface area in total (~100 m2) for performing efficient gas exchange.
The alveoli form a honeycomb of cells around the spiral, cylindrical surface of the alveolar duct. The exposed alveolar surface is normally covered with a surface film of lipoprotein material.
There are several types of pulmonary alveolar cells. Type I (or small type A), are non-phagocytic, membranous pneumocytes. These surface-lining epithelial cells are approximately 5 μm in thickness and possess thin squamous cytoplasmic extensions that originate from a central nucleated portion. These portions do not have any organelles and hence they are metabolically dependent on the central portion of the cell. This reduces their ability to repair themselves if damaged. Attached to the basement membrane are the larger alveolar cells (Type II, type B or septal cells). These rounded, granular, epithelial pneumocytes are approximately 10 to 15 μm tick. There are 6 to 7 cells per alveolus and these cells possess great metabolic activity. They are believed to produce the surfactant material that lines the lung and to be essential for alveolar repair after damage from viruses or chemical agents.
Amongst, the important roles of the lungs, one can cite: (i) supply oxygen, (ii) remove wastes and toxins, and (iii) defend against hostile intruders. The lungs have three dozen distinct types of cells. Some of these cells scavenge foreign matter. Others have cilia that sweep the mucous membranes lining the smallest air passages. Some cells act on blood pressure control, while others spot infection invaders.
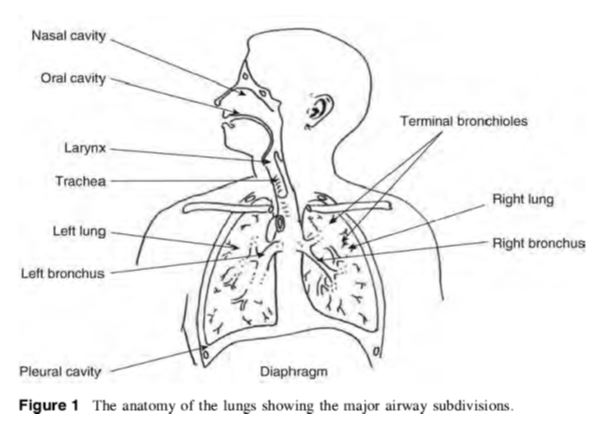
The respiratory system is susceptible to a number of diseases, and the lungs are prone to a wide range of disorders caused by genetic factors, infection and pollutants in the air. The most common problems of the respiratory system are:
- Asthma
- Bronchiolitis
- Chronic obstructive pulmonary disease (COPD)
- Common cold
- Cough
- Cystic fibrosis (CF)
- Lung cancer
- Pneumonia
- Pulmonary hypertension
PRINCIPAL MECHANISMS OF RESPIRATORY DEPOSITION
The deposition of inhaled particles in the different regions of the respiratory system is very complex, and depends on many factors. Some of the factors influencing respiratory deposition include:
- Breathing rate
- Mouth or nose breathing
- Lung volume
- Respiration volume
- Health of the individual
- Bifurcations in the airways result in a constantly changing hydrodynamic flow field.
Depending on the particle size, airflow, and location in the respiratory system, particle deposition occurs via on of the following principal mechanisms:
Impaction
Each time the airflow changes due to a bifurcation in the airways, the suspended particles tend to travel along their original path due to inertia and may impact on an airway surface. This mechanism is highly dependent on aerodynamic diameter, since the stopping distance for very small particles is quite low. Impaction occurs mostly in the case of larger particles that are very close to airway walls, near the first airway bifurcations. Therefore, deposition by impaction is greatest in the bronchial region. Impaction accounts for the majority of particle deposition on a mass basis.
Sedimentation
Sedimentation is the settling out of particles in the smaller airways of the bronchioles and alveoli, where the air flow is low and airway dimensions are small. The rate of sedimentation is dependent on the terminal settling velocity of the particles, so sedimentation plays a greater role in the deposition of particles with larger aerodynamic diameters. Hygroscopic particles may grow in size as they pass through the warm, humid air passages, thus increasing the probability of deposition by sedimentation.
Interception
Interception occurs when a particle contacts an airway surface due to its physical size or shape. Unlike impaction, particles that are deposited by interception do not deviate from their air streamlines. Interception is most likely to occur in small airways or when the air streamline is close to an airway wall. Interception is most significant for fibers, which easily contact airway surfaces do to their length. Furthermore, fibers have small aerodynamic diameters relative to their size, so they can often reach the smallest airways.
Diffusion
Diffusion is the primary mechanism of deposition for particles less than 0.5 microns in diameter and is governed by geometric rather than aerodynamic size. Diffusion is the net transport of particles from a region of high concentration to a region of lower concentration due to Brownian motion. Brownian motion is the random wiggling motion of a particle due to the constant bombardment of air molecules. Diffusional deposition occurs mostly when the particles have just entered the nasopharynx, and is also most likely to occur in the smaller airways of the pulmonary (alveolar) region, where air flow is low.
Absorption – bioavailability of drugs
Although inhaled drugs have been used for over 50 years to treat airway disease and are in development or being considered for the treatment of many other lung diseases, insulin is at present time the only one representative inhaled drug on the market for systemic disease. Exubera® (insulin human [rDNA origin] inhalation powder is the first diabetes treatment which can be inhaled. Exubera® helps control high blood sugar, works in adults with type 1 diabetes and with type 2 diabetes as well This therapeutic success has lead a number of other companies to investigate and to advance clinical trials as inhaled formulations for systemic applications with a variety of large molecules (leuprolide, a luteinizing hormone-releasing hormone (LHRH) analogue, …). Recent advances in the development of particle technologies and devices now make it possible to formulate, stabilize, and accurately deliver almost any drug to the lungs.
The pulmonary membrane is naturally permeable to small molecule drugs and to many therapeutic peptides and proteins. The epithelium of the lung, the significant barrier to absorption of inhaled drugs, is thick (50–60 μm) in the trachea, but diminishes in thickness to an extremely thin 0.2 μm in the alveoli. The change in cell types and morphology going from trachea, bronchi, and bronchioles to alveoli is very dramatic. The lungs are for more permeable to macromolecules than any other portal of entry into the body. Some of the most promising therapeutic agents are peptides and proteins, which could be inhaled instead of injected, thereby improving compliance .Particularly, peptides that have been chemically altered to inhibit peptidase enzymes exhibit very high bioavailabilities by the pulmonary route .Indeed, natural mammalian peptides, les than 30 amino acids (somatostatin, vaso active intestinal peptide [VIP], and glucagons), are broken down in the lung by ubiquitous peptidases and have very poor bioavailabilities. Conversely, proteins with molecular weights between 6000 and 50,000 Da are relatively resistant to most peptidases and have good bioavailabilities following inhalation. For larger proteins, the bioavailabilities and absorption mechanisms are not well completely elucidated.
ADVANTAGES OF PULMONARY DRUG DELIVERY SYSTEM
- The ability to nebulize viscous drug formulations for pulmonary delivery, thereby overcoming drug solubility issues with the ability to use lipid, water or lipid/water emulsions as drug carriers.
- Ability to nebulize viscous liquids into droplets in the 2-5μm range regardless of the carrier composition solubility which would allow for a wide range of drug formulation options.
- Increased drug delivery efficacy due to size-stable aerosol droplets with reduced
hygroscopic growth and evaporative shrinkage.
- Liposomal drug formulations remain stable when nebulized.
- Ability to nebulize protein-containing solutions.
- For hand held inhaler applications, drug does not need to be emulsified in liquefied nebulizing gas to achieve aerosolization.
![[PPT PDF] Pharmaceutical Water System Validation - IDENTIFICATION OF MICROORGANISMS](https://pharmawiki.in/wp-content/uploads/2017/11/PPT-PDF-Pharmaceutical-Water-System-Validation-IDENTIFICATION-OF-MICROORGANISMS.jpg)
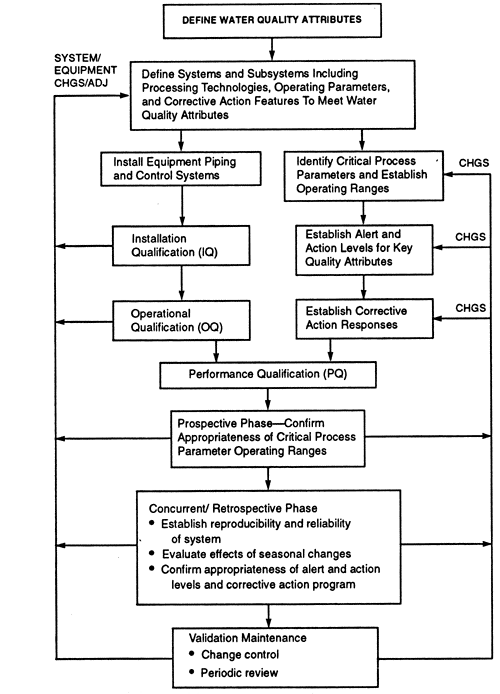
![[PPT PDF] Pharmaceutical Water System Design Validation -UNIT OPERATIONS CONCERNS im](https://pharmawiki.in/wp-content/uploads/2017/11/PPT-PDF-Pharmaceutical-Water-System-Design-Validation-UNIT-OPERATIONS-CONCERNS.jpg)
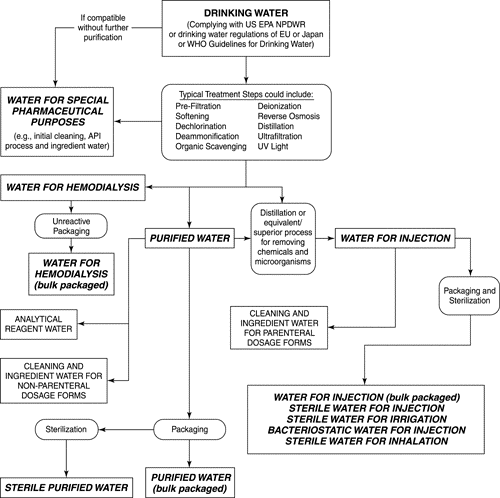
![[PPT PDF] Pharmaceutical Water System Design Validation -SAMPLING CONSIDERATIONS](https://pharmawiki.in/wp-content/uploads/2017/11/PPT-PDF-Pharmaceutical-Water-System-Design-Validation-SAMPLING-CONSIDERATIONS.jpg)
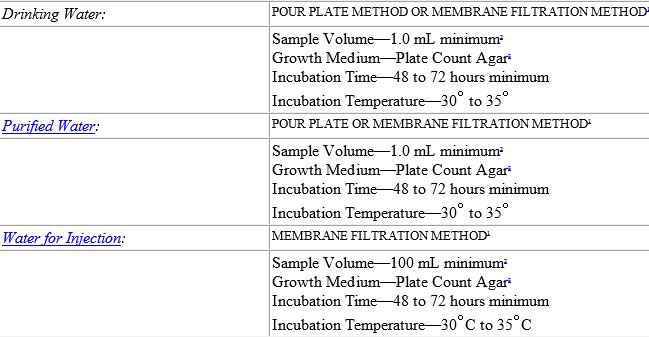
![[PPT PDF] Pharmaceutical Water System Design Validation - Microbial Testing of Water](https://pharmawiki.in/wp-content/uploads/2017/11/PPT-PDF-Pharmaceutical-Water-System-Design-Validation-Microbial-Testing-of-Water.jpg)